Substituční reakce
Pro aromatické uhlovodíky jsou typické elektrofilní substituční reakce.
Mechanismus elektrofilní substituce můžeme rozložit do několika stupňů:
-
rychlá interakce (adice) elektrofilního činidla s delokalizovanými π–elektrony, při které vzniká útvar, často označovaný jako π–komplex,
-
pomalý přechod π komplexu na strukturně definovaný σ–komplex s dočasně porušenou aromaticitou,
-
odštěpení protonu z atomu uhlíku, k němuž je vázán elektrofil, obnovení aromatického charakteru, vznik substitučního derivátu.
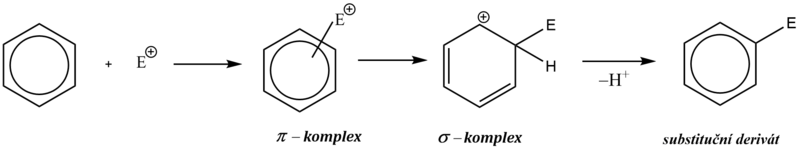
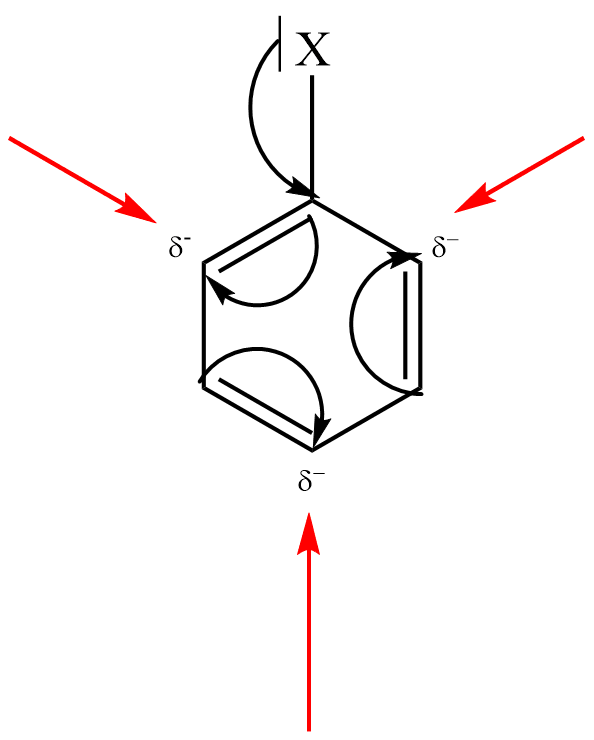
Elektrofilní substituce je zahájena adicí elektrofilní částice na aromatický uhlovodík, kdy elektrofil je zachycen π-elektronovým oblakem aromatického systému (aromatický systém vykazuje zvýšenou elektronovou hustotu). Vzniká útvar označovaný jako π–komplex.
π–komplex poté pomalu přechází na tzv. σ–komplex, v němž elektrofil nahradil jeden z vodíkových kationtů na benzenovém jádře a je již vázán na konkrétní atom uhlíku.
σ–komplex má povahu kationtu a nemá již aromatický charakter, protože překryv 2p orbitalů je znemožněn přítomností atomu uhlíku v hybridním stavu sp3 (uhlík s navázaným elektrofilem).
Kladný náboj σ–komplexu je ovšem stabilizován konjugovaným systémem dvojných vazeb. Tento fakt se často zdůrazňuje grafickým zápisem s neúplným kruhem, znázorňujícím konjugaci dvojných vazeb.
V závěrečné fázi reakce dochází k odštěpení protonu z atomu uhlíku, k němuž je vázán elektrofil, čímž dojde k obnově aromatického charakteru. Obnovení aromatického systému je obecně hnací silou celé aromatické elektrofilní substituce.
Protože všechny vodíkové atomy v benzenu jsou rovnocenné, má reakce u benzenu jednoznačný průběh a může vznikat pouze jediný produkt substituce. Pokud však uvažujeme již substituovaný benzenový derivát, budou produkty záviset na charakteru substituentů, které byly na aromatickém jádře navázány před provedením reakce.
Mezi nejvýznamnější reakce probíhající tímto mechanismem patří halogenace, sulfonace, nitrace, alkylace a acylace. Pokud není činidlo již samo o sobě dostatečně polární, je nutné vytvořit podmínky pro vznik elektrofilní částice. Pro tento účel se běžně využívají Lewisovy kyseliny, jako např. chlorid hlinitý.
1. Halogenace (chlorace, bromace, výjimečně jodace)
Praktický význam mají pouze přímé chlorace a bromace. Fluor je příliš reaktivní a jod zase příliš málo reaktivní.
Chlorace i bromace se musí provádět v přítomnosti Lewisových kyselin (AlCl3 , FeCl3 , FeBr3 ), které působí jako katalyzátor reakce.
Elektrofilní částice, kterou je kation halogenu, se totiž tvoří heterolytickým štěpením molekuly halogenu účinkem Lewisovy kyseliny – nejčastěji halogenidu železa nebo hliníku podle rovnice:
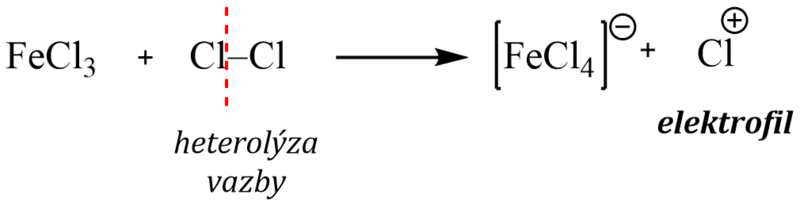
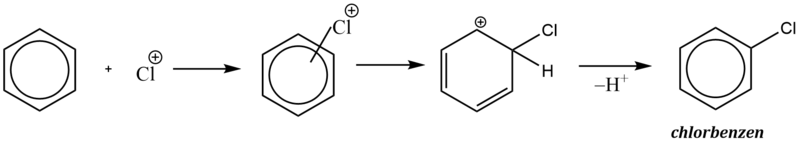
Komplexní kation [FeCl4]- vytrhává ze σ–komplexu proton a rozpadá se na chlorovodík a katalyzátor:

2. Sulfonace
Vodík na aromatickém jádře se nahrazuje sulfoskupinou –SO3H za vzniku arensulfonové kyseliny. Jako sulfonační činidlo se používá koncentrovaná kyselina sírová, oleum (roztok oxidu sírového rozpuštěného v kyselině sírové), popř. oxid sírový. Elektrofilem je kation SO3H+, vzniklý protonizací kyseliny sírové a následným odštěpením vody:
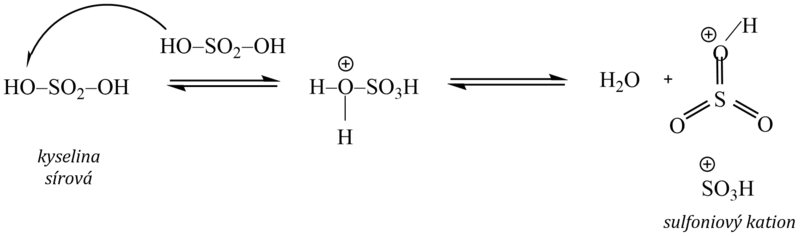
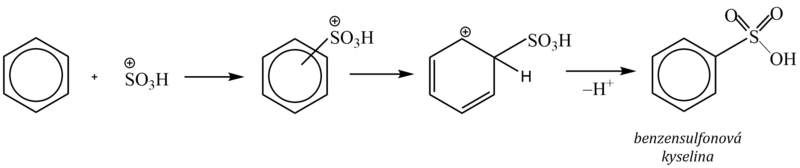
Sulfonace je jedinou elektrofilní substitucí, která je vratná, neboť účinkem koncentrované kyseliny sírové (nebo olea) sulfoskupinu do molekuly aromátu vnášíme, při použití zředěné kyseliny sírové sulfoskupinu naopak odstraníme.
3. Nitrace
Nitrací zavádíme do molekuly arenů skupinu NO2. Při reakci vznikají aromatické nitrosloučeniny, které jsou významnými sloučeninami jakožto meziprodukty dalších syntéz.
Jako nitrační činidlo se používá směs kyseliny sírové a dusičné (tzv. nitrační směs), ve které dochází působením kyseliny sírové k protonizaci kyseliny dusičné a následnému vzniku nitroniového kationtu, který je vlastní elektrofilní částicí. Kyselina sírová tedy slouží jako zdroj protonů a zároveň má funkci dehydratačního činidla (reakční voda by způsobila disociaci kyseliny dusičné a nitrace by se zastavila).
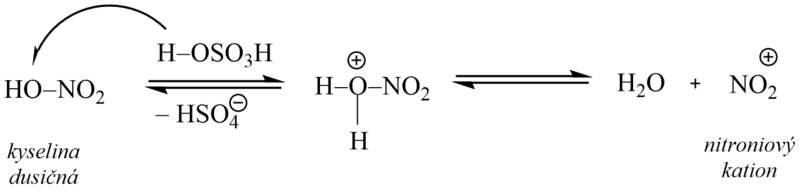
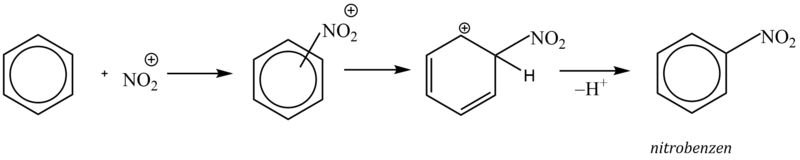
4. Friedel – Craftsova alkylace
Alkylačními reakcemi zavádíme do molekuly arenů alkylskupinu (alifatické uhlovodíkové zbytky). Jako alkylační činidla se proto používají alkylhalogenidy (nejčastěji alkylchloridy nebo bromidy). Reakci lze uskutečnit pouze v přítomnosti katalyzátoru, Lewisovy kyseliny. Nejčastěji používaným katalyzátorem je bezvodý chlorid hlinitý AlCl3 .
K alkylacím je možno také použít alkeny nebo alkoholy, reakce je však v tomto případě katalyzovaná kyselinami.
Elektrofilní činidlo může proto vznikat těmito reakcemi:
1)
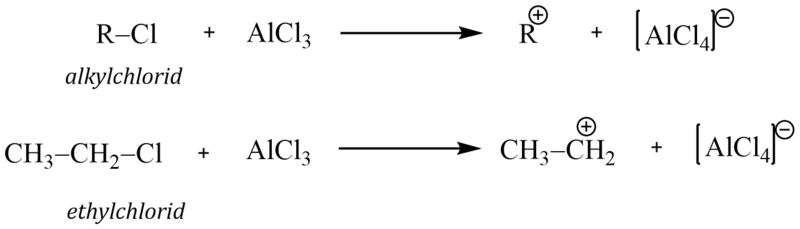
2)
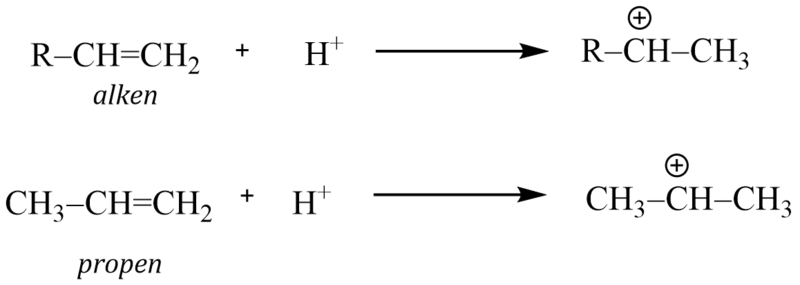
3)

Jako příklad si uvedeme syntézu ethylbenzenu. Reakční mechanismus zahrnuje reakci Lewisovy kyseliny s alkylhalogenidem za vzniku karbokationtu, který pak jako elektrofilní činidlo napadá aromatický systém:
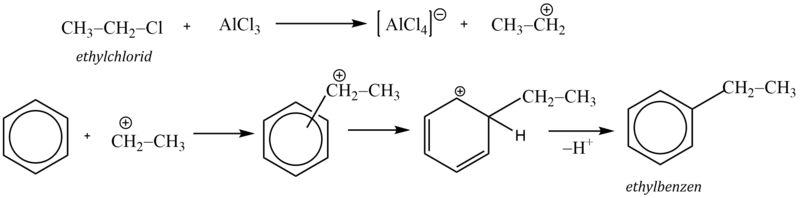
Komplexní kation [AlCl4]- vytrhává ze σ–komplexu proton a rozpadá se na chlorovodík a katalyzátor:

Při použití alkenu jako alkylačního činidla vzniká na aromátu více rozvětvený alkyl.
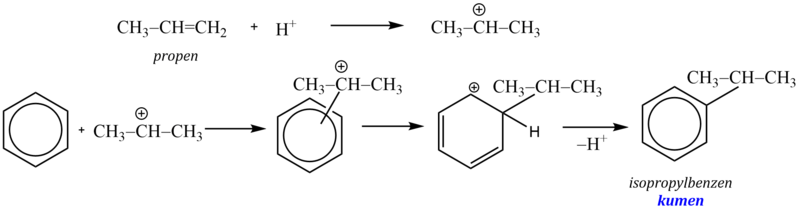
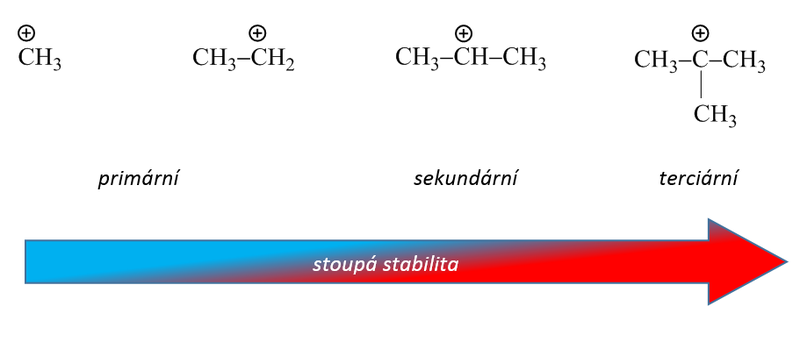
Snadnost alkylace při použití alkylhalogenidů závisí na stabilitě vznikajícího karbokationtu (terciární > sekundární > primární). Vzniká-li při reakci s katalyzátorem primární kation pak, je-li to možné s ohledem na počet uhlíkových atomů v řetězci, dojde k izomeraci (přesmyku) za vzniku stabilnějšího meziproduktu. Proto nelze zavést do aromátu delší nerozvětvené alkylové skupiny, protože spontánně izomerizují za vzniku stabilnějších sekundárních a terciárních karbokationtů.
Příkladem může být reakce 1-chlorpropanu s benzenem v přítomnosti chloridu hlinitého, při které vzniká směs isopropylbenzenu a propylbenzenu v poměru 1:1. Příčinou je přesmyk primárního kationtu na stabilnější sekundární:
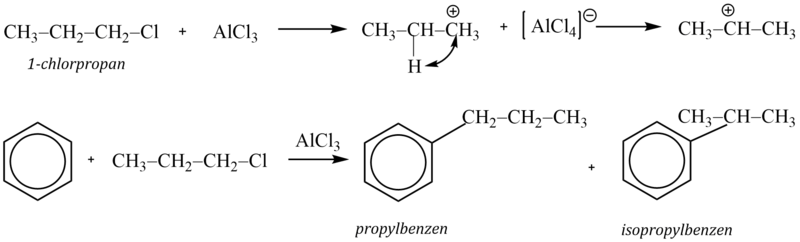
Alkylace se však setkává s celou řadou problémů. Alkylskupina usnadňuje další alkylaci (vyvolává +I efekt), a tudíž vzniklý alkylaren může být ještě reaktivnější než výchozí látka. V důsledku toho dochází k následným alkylacím. K nim dochází i při použití stechiometrického poměru aren-alkylační činidlo 1:1.
5. Friedel – Craftsova acylace
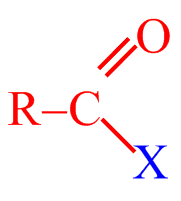
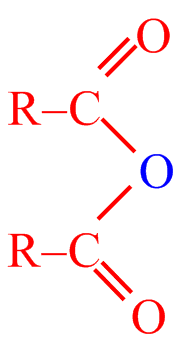
Acylačními reakcemi se do molekuly arenů zavádí acylová skupina R–CO–. Při reakci tak vznikají alkylarylketony. Jako acylační činidla se používají funkční deriváty karboxylových kyselin, jako acylhalogenidy (RCOX), anhydridy ((RCO)2 O) nebo samotné karboxylové kyseliny (RCOOH).
Jako katalyzátory se používají Lewisovy kyseliny (AlX3).
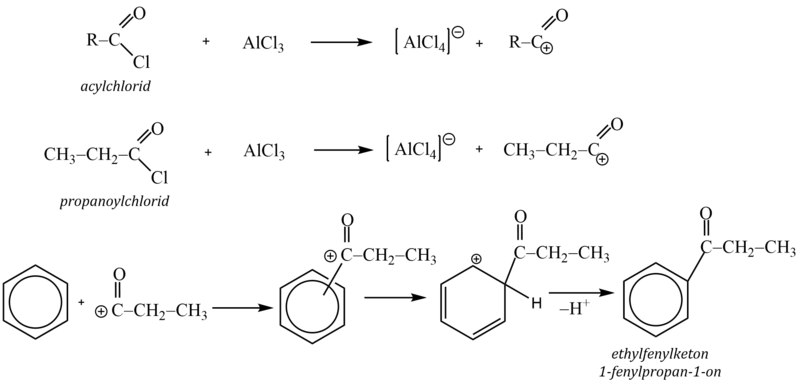
Protože u acylací nedochází k izomeracím, lze Friedelovy-Craftsovy acylace využít k zavedení lineárního alkylového řetězce do aromatického systému.
Vliv substituentů na průběh další substituce u derivátů benzenu
Substituent navázaný na benzenové jádro působí na toto jádro svým elektronovým efektem, čímž mění elektronovou hustotu na jednotlivých uhlících, a ovlivňuje tak vstup dalšího substituentu na benzenové jádro. Jaké produkty budou vznikat, závisí proto na charakteru substituentů, které byly na aromatickém jádře navázány před provedením reakce (např. u toluenu je to methylskupina).
Podle toho se substituenty dělí do dvou tříd:
-
substituenty I. třídy (orientují další substituci do polohy o- a p-),
-
substituenty II. třídy (orientují další substituci do polohy m-).
1. Substituenty I. třídy (aktivující substituenty)
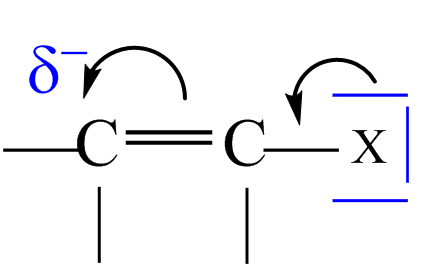
Tyto substituenty vykazují vůči benzenovému jádru kladný mezomerní efekt +M efekt, resp. kladný indukční + I efekt, tzn. že mají volný elektronový pár na atomu, který je vázán na jádro, a ten poskytují do konjugace s π–elektrony arenu, a tím další substituci usnadňují. Substituenty proto řídí další substituci přednostně do poloh ortho- a para- .
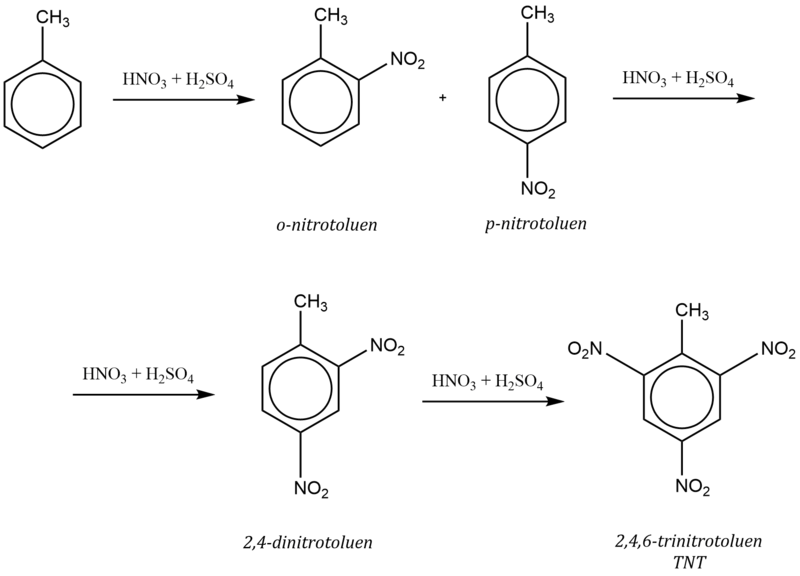
Obecně patří do této skupiny substituenty obsahující volný elektronový pár (–NH2 , –OH, –OR, –NR2 , –NHR, –Ar, –R, halogeny …) a uhlovodíkové zbytky .
Např. nitrace toluenu probíhá podle schématu:
2. Substituenty II. třídy (desaktivující substituenty )
Tyto substituenty vykazují vůči jádru záporný mezomerní efekt –M, tzn. že obsahují atomy elektronegativnějšího prvku vázané dvojnou nebo trojnou vazbou, a proto odčerpávají z arenů π–elektrony, a tím znesnadňují další substituci. Substituenty proto řídí další substituci do polohy meta–.
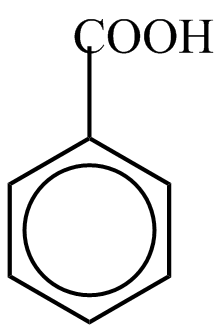
Do této skupiny patří substituenty: –NO2 , –C ≡ N, –CH=O, –SO3H, –COOH…
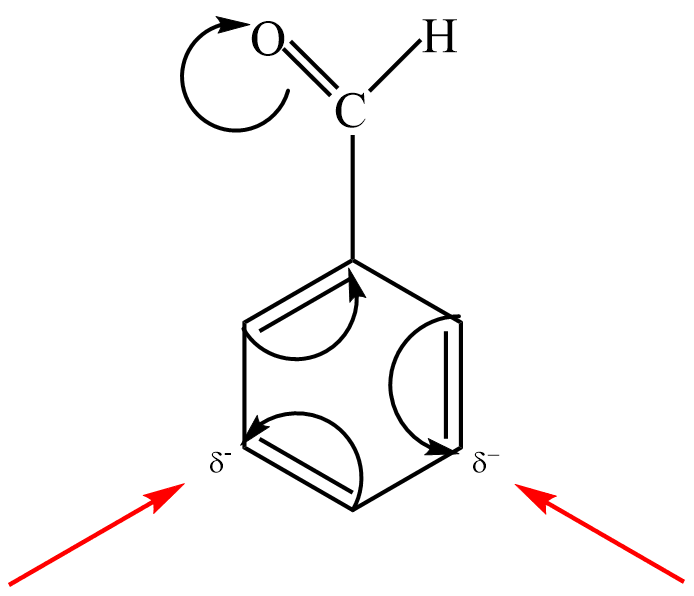
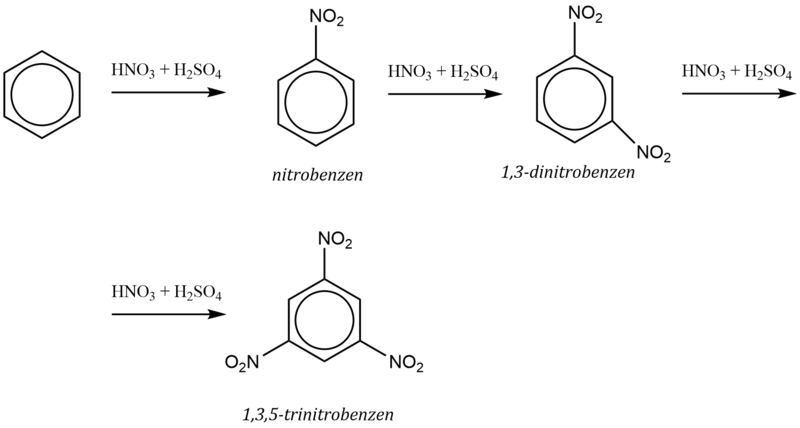
Např. nitrace benzenu do druhého stupně probíhá dle schématu:
Pokud jsou na benzenu dva, popř. více substituentů, je reaktivita benzenového jádra ovlivněna efekty všech přítomných substituentů. Ve většině případů lze snadno předpovědět, do jaké polohy se další substituent umístí. Přitom je nutné vedle elektronových efektů přihlédnout i ke stérickým faktorům (objemnosti substituentů).
Vliv substituentů na průběh další substituce u naftalenu
Na rozdíl od benzenu v naftalenu již nejsou všechny atomy uhlíku rovnocenné. Elektrony π již nejsou zcela rovnoměrně rozložené a důsledkem částečné lokalizace je relativně vyšší elektronová hustota v polohách α (1, 4, 5, 8). Proto elektrofilní substituce, jejíž mechanismus je obdobný jako u benzenu, probíhá přednostně do polohy α.
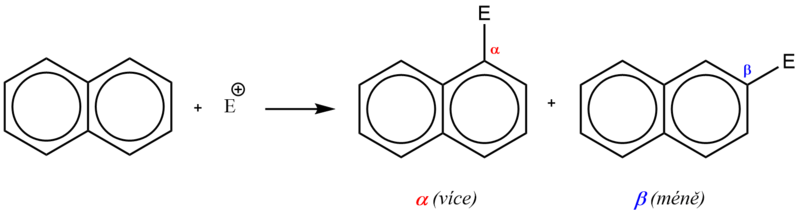
U monosubstituovaného naftalenu je vstup dalšího substituentu orientován v závislosti na charakteru a poloze přítomného substituentu.
1. Substituenty I. třídy v poloze α
Substituenty I. třídy v poloze 1 (α) (donory elektronů, aktivující substituenty) usměrňují další substituci do téhož jádra do poloh 2 a 4.
2. Substituenty I. třídy v poloze β
Nachází-li se substituent v poloze 2 (β), pak orientuje vstup dalšího substituentu do polohy 1 na témže jádru.
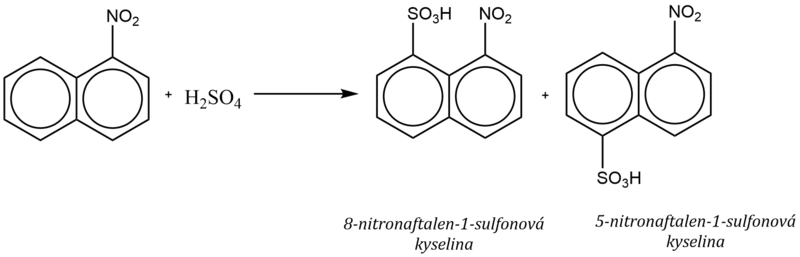
3. Substituenty II. třídy v poloze α nebo β
Substituenty II. třídy (deaktivující substituenty) snižují π–elektronovou hustotu na aromatickém jádře, na kterém jsou vázané, což se projevuje tím, že další substituce proběhne na sousedním jádře do poloh 5 a 8, bez ohledu na polohu řídícího substituentu.
- FIKR, Jaroslav a Jaroslav KAHOVEC. Názvosloví organické chemie. Olomouc: Rubico, 2002, ISBN 80-7346-017-3.
- HONZA, Jaroslav a Aleš MAREČEK. Chemie pro čtyřletá gymnázia 2. díl. Olomouc: Nakladatelství Olomouc, 2005, ISBN 80-7182-141-1.
- JANECZKOVÁ, Anna a Pavel KLOUDA. Organická chemie. Ostrava: Nakladatelství Pavel Klouda, 1998, ISBN 80-902155-6-4/9802.
- KOTLÍK, Bohumír a Květoslava RŮŽIČKOVÁ. Chemie v kostce II.. Havlíčkův Brod: Fragment, 1997, ISBN 80-7200-342-9.
- PACÁK, Josef. Jak porozumět organické chemii. Praha: Karolinum, 1997, ISBN 80-7184-261-3.
- SVOBODA, Jiří a kol. Organická chemie I. Praha: Vysoká škola chemicko-technologická, 2005, ISBN 80-7080-561-7.
Obrázky
- Obr. 1: Autor neznámý. www.wikiwand.com [online]. [cit. 26.10.2014]. Dostupný na WWW: http://upload.wikimedia.org/wikipedia/commons/thumb/8/87/Hueckel.jpg/220px-Hueckel.jpg
- Obr. 2: Autor neznámý. www.wikiwand.com [online]. [cit. 26.10.2014]. Dostupný na WWW: http://upload.wikimedia.org/wikipedia/en/2/21/Gilbert_N_Lewis.jpg
Pokud není uvedeno jinak, autorem obrázků je Mgr. Michal Bezděk.